
Chromosome no21
The idea for this project was spawned from a random conversation with a classmate of mine. The topic was, “How big would an image that uses one pixel for one base pair for the entire human genome be?” The answer to that question is, for the curious, around 14.7 x 14.7 meters on your typical 96 dpi monitor. But this sort of visualization tends to look like this — high-contrast and eye-straining, with little pattern discernible. In fact, there are two choices that make the linked visualization less-than-optimal. One, even though Adenine/Thymine and Guanine/Cytosine are quite related in the grand scheme of things, the drastically different coloring of them makes relating them hard. Two, because the pixels are placed horizontally, two pixels that are adjacent are not very close to each other at all. Related to this point, the ‘interesting patterns’ that appear are entirely arbitrary, depending on the width of the image and the strain it puts on the human eye.
I remember a task on Rosalind that had you count the ratio of G and C to the total. Rosalind is a very recommendable website that introduces a beginner programmer to string-related problems from the real world. In fact, most of the tasks on Rosalind are actual ones encountered in biometrics. I decided that grouping up a number of base pairs and representing them as a single pixel, shaded depending on its GC content would be better. In fact, areas with high GC content(called GC islands) are very helpful in annotating genes.
The shading itself is also an issue. Simple RGB interpolating is rather bad. RGB interpolation tends to be unintuitive compared to what you’d expect. The default gradient displayed here shows that colors dim and grey between purple and green. The Lab scheme I ended up using also still has problems in blending (colors lose saturation), but it also has a great advantage over Lch and RGB, in that it’s possible to construct linear gradients that start as one color, become white in the middle, then become another color at the end. In my opinion, this is a great feature of Lab that justifies using it over the superior Lch color space.
The raw data for the human genome was pulled from the Human Genome Project, hosted at Project Gutenburg.
First, I need to count the base pairs in a given chromosome’s sequence file. Looking at chromosome 21’s file(WARNING: VERY LARGE FILE, 46.7 MB), chosen because it’s the smallest chromosome, there are about 378 lines of preamble before the actual sequence starts. The first line of the sequence is leaded by the following text:
>chr21 Homo sapiens chromosome 21
Hmm–this gives me a hint on how to start processing the text. I take a look at the Y chromosome’s file, too (found here, also very large at 70.2 MB), and find the line
>chrY
Both lines are at line 379 of both files, but I decide to use “a greater-than sign followed by text” as the starting point of nucleotide processing. This also provides a handy way to name the resulting file–split the line with a > by spaces then remove the > of the first split string.
aligned = False
outname = ""
counter = 0
for line in file:
counter += 1
if counter % 10000 == 0:
print counter
if not aligned:
if line[0] == ">":
txts = line.split(" ")
print "Checked: " + txts[0]
outname = txts[0].strip()[1:]
aligned = True
That explains this section of the script. The variable aligned is to enable my program to switch between “scan file for start of nucleotide sequence” mode and “process nucleotides” mode; counter was added later to show progress instead of a supposedly frozen console.
Next, I look at the bulk of the file (chr21 is 898531 lines long!). Each line has at most 50 chromosomes on it. I want to let the script use an arbitrary number of base pairs per pixel, so I code a ‘buffer’ that, when full, generates the data for a pixel then empties.
else:
group += line.strip()
if len(group) >= pixelsize:
GC = AT = N = 0
working = group[:pixelsize] # Copy the base pairs to work on.
group = group[pixelsize:] # Remove the worked-on pairs.
for dna in working:
if dna == 'A' or dna == 'T':
AT += 1
elif dna == 'G' or dna == 'C':
GC += 1
elif dna == 'N':
N += 1
else:
unknowns.append(dna)
info = []
if AT+GC == 0:
info.append(0.0)
else:
info.append(GC/float(AT+GC))
if N != 0:
info.append(N/float(pixelsize))
ratios.append(info)
Something very noticable in chr21 is that the first couple 225 thousand lines are all
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
This isn’t in the five nucleotides most people know of, which are Adenine, Thymine, Guanine, Cytosine, and Uracil. The answer to this problem (which took me somewhere between ten and thirty minutes to figure out, due to ‘N’ being an extremely common letter) can be found here. Basically, N represents nucleotides that differ from person to person, as the Human Genome Project sequenced multiple unique genomes to produce its final report. In my visualization, Ns are represented by interpolating between black and whatever color the pixel is after considering GC content.
Using the Python library Image to plot the data linearly (that is, left-to-right, top-to-down in order) looks like this:

Like I mentioned above, this is not a very good visualization. Pixels are tiny, and the difference between two vertically adjacent ones is on the order of 47400 base pairs. To remedy this, I use the Hilbert curve.

From Wikipedia
The Hilbert curve is a space-filling curve, and makes sure adjacent points are also fairly close to each other along the length of the curve.

From Wikipedia
The difference between two adjacent points because exponentially larger depending on which ‘square’ the two points are in, as illustrated in the above image. This happens because the Hilbert curve is a recursive, fractal curve, but is still a great improvement over the linear layout where every line is a huge jump.
The code I use to generate a list of Hilbert points is borrowed from Malcolm Kesson at http://www.fundza.com, with slight edits to store coordinates in a list rather than print to the screen.
def hilbert(x0, y0, xi, xj, yi, yj, n, coords):
if n <= 0:
X = x0 + (xi + yi)/2
Y = y0 + (xj + yj)/2
# Output the coordinates the cv
coords.append((X,Y))
else:
hilbert(x0, y0, yi/2, yj/2, xi/2, xj/2, n - 1, coords)
hilbert(x0 + xi/2, y0 + xj/2, xi/2, xj/2, yi/2, yj/2, n - 1, coords)
hilbert(x0 + xi/2 + yi/2, y0 + xj/2 + yj/2, xi/2, xj/2, yi/2, yj/2, n - 1, coords)
hilbert(x0 + xi/2 + yi, y0 + xj/2 + yj, -yi/2,-yj/2,-xi/2,-xj/2, n - 1, coords)
A 0^th order Hilbert curve has 1 point; a 1^st order Hilbert curve has 4 points, a 2^nd order Hilbert curve has 16 points, and so on. To determine how many points are needed–that is, what N in N^th order Hilbert curve I need, I use simple math.
powers = int(math.ceil(math.log(math.sqrt(len(ratios)))/math.log(2)))
size = 2**powers
… Perhaps not so simple. Here it is using Latex markup:

L is the length of the current chromosome. I take the square root of it to find the length of a square that would have area of L, then take the logarithm of that and ceil() it to find n so that 2^n has an area of at least L.
Now we have a list of points, ordered so that if they are popped from the front and a line drawn they would draw a Hilbert curve. Next up is actually drawing the pixels.
exceptions = 0
for d in xrange(len(ratios)):
if d % 10000 == 0:
print d,"/",len(ratios)
if mode == "lab":
col = lerpLab(ATColor, GCColor, ratios[d][0])
else:
col = lerpLch(ATColor, GCColor, ratios[d][0])
if len(ratios[d]) > 1:
if mode == "lab":
col = lerpLab(col, black, ratios[d][1])
else:
col = lerpLch(col, black, ratios[d][1])
# translate Lab/Lch to RGB colors
r,g,b = conversions.convert_color(col, colors.sRGBColor).get_value_tuple()
# Some Lab/Lch colors can't be represented using RGB.
if r < 0.0:
r = 0.0
if g < 0.0:
g = 0.0
if b < 0.0:
b = 0.0
if r > 1.0:
r = 1.0
if g > 1.0:
g = 1.0
if b > 1.0:
b = 1.0
x, y = coords[d]
try:
pixels[x,y] = (int(r*255),int(g*255),int(b*255), 255)
except IndexError:
print x,y
exceptions+=1
exceptions is a counter of out-of-bounds exceptions, which I had some trouble with before realizing that Hilbert curves needed to have squares that are 2^n in side length to be all integers. Again, I print the current progress along with the total number of things to process (which I now know, having read in the whole file), and finally decide the color of each pixel.
lerpLab and lerpLch are simple functions that interpolate linearly between the two given colors by t, so that when t=0 the result is 100% Color1, and when t=1 the result is 100% Color2. I use colormath for Lab color calculations and converting to RGB.
img.save("output/" + outname + ".png")
And the image is produced.

Chromosome no21
Areas that are close on the image tend to be close on the actual chromosome, too. Blue areas are also more visible than the linear version, indicating the presence of GC islands, which are typically 4 blue spots together.
I plan on making a slightly more interactive version of the image with Javascript, to display the sequence at a pixel when clicked, in order to let the user search for the sequence using BLAST].
Repository here.














 and isFixed.
and isFixed.  , mass,
, mass, 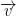 , I (which is the moment of inertia for a thin, evenly dense rod). It also has functions to apply force to the center of mass (
, I (which is the moment of inertia for a thin, evenly dense rod). It also has functions to apply force to the center of mass (
 is a position vector from node1 to the center of mass;
is a position vector from node1 to the center of mass;  is a formula for torque.
is a formula for torque.

 . Timestep is usually [0.0001, 0.001]; larger time steps tend to make the system look too stuttery, and smaller steps make the simulation look like it is moving in slow motion.
. Timestep is usually [0.0001, 0.001]; larger time steps tend to make the system look too stuttery, and smaller steps make the simulation look like it is moving in slow motion.

